8 Mainstream Synthetic Accessibility Prediction Models: A Technical Comparison
If you spent an entire week having computationally generated a million beautiful new molecules, and the next morning you still face the million-dollar question - "How many of them can actually be made?"
You'd probably stare at the screen and not want to answer.
We understand that silence.
Computational tools can generate molecular structures fast - but the flasks and reagents in the lab don't care about what computers promise on paper. Synthetic accessibility is the bottleneck choking many drug design projects.
To address this, the Synthetic Accessibility Score (SA Score) has become a core tool. Below, we walk through 8 mainstream synthetic accessibility prediction models - their technical principles, strengths, and the scenarios each is best suited for.
Three Technical Approaches
The field currently follows three main technical routes for synthetic accessibility prediction.
1. Heuristic (Rule-Based) Methods - Rely on expert chemical knowledge and manually defined scoring functions to estimate synthesis difficulty.
2. Machine Learning / Data-Driven Methods - Train statistical or deep learning models on large-scale chemical reaction datasets to automatically learn the patterns of synthesis.
3. Retrosynthesis-Based Methods - Directly simulate real synthesis pathways using retrosynthetic analysis to determine whether a molecule can actually be made.
SAScore: Scoring by "Familiarity"
Developed in 2009 by a team at the Novartis Institutes for BioMedical Research,1 SAScore works along two dimensions:
Molecular complexity - calculated from molecular weight, number and type of ring systems, stereocenters, and other topological features that directly reflect structural complexity.
Fragment familiarity - one million molecules from PubChem are decomposed into substructural fragments following standard rules, and each fragment frequency in large chemical databases is recorded. Common fragments add points; rare fragments subtract points.
The final SAScore maps to a scale of 1 to 10, where 1 means extremely easy to synthesize, and 10 means extremely difficult. This allows direct comparison across molecules.
Strengths: Millisecond-level computation per molecule. Intuitive logic that medicinal chemists understand and trust. Deployable without heavy computational infrastructure.
Limitations: Cannot clearly distinguish "structurally complex" from "synthetically difficult." Molecules with elaborate structures, but well-established synthetic routes, may be scored too harshly. Scores are based on static structural features and do not account for actual reaction feasibility.
BR-SAScore: A Patch for SAScore
BR-SAScore largely preserves the SAScore framework but precisely addresses its key blind spot - no consideration of actual chemical reaction feasibility.2
Its core upgrade is a fragment-source scoring mechanism. It distinguishes between "fragments from commercially available building block libraries" and "fragments generated through chemical reactions," encoding reaction rules and building block knowledge from the retrosynthesis software into computable chemical fingerprint scores. This effectively creates a lightweight CASP surrogate. The result is a score that better reflects the intuition of an experienced synthetic chemist, rather than just static structural features.
Strengths: Directly addresses SAScore's core blind spot by incorporating real reaction feasibility into the scoring function. Retains SAScore's speed and interpretability while meaningfully improving accuracy. Because commercial building block availability is encoded directly into the score, results are more actionable for chemists planning actual synthesis.
Limitations: Still fundamentally a heuristic model that does not simulate actual reaction pathways. Performance is tied to the coverage and currency of the commercial building block library used. For highly novel scaffolds that fall outside the reaction rules, it offers limited improvement over the original SAScore.
SYBA: A Bayesian Probability Game
SYBA (SYnthetic Bayesian Accessibility) is a synthetic accessibility prediction model built on Bayesian probability theory.3 It answers one straightforward question: do the local fragments of a molecule appear more often in easy-to-synthesize (ES) molecules, or in hard-to-synthesize (HS) ones?
SYBA computes the frequency of ECFP4 molecular fingerprint fragments in ES and HS molecular libraries, then uses those frequency differences to judge accessibility. Each fragment receives an "ease-of-synthesis tendency" score, and the scores are summed to produce a final prediction.
Strengths: Interpretable - you can see each fragment's contribution and identify exactly where the difficulty comes from.
Limitations: Assumes fragments are independent of each other, ignoring the actual difficulty of forming bonds between them. The hard-to-synthesize training samples are algorithmically generated, and the quality of those samples directly determines model accuracy.
SCScore: Learning from Reaction Data
SCScore (Synthetic Complexity Score) marks a turning point.4 Rather than relying on expert-defined rules, it learns from large-scale real reaction data.
It operates on one iron law: the complexity of a product must be greater than or equal to the complexity of its reactants. Using this "directionality constraint," it learns what structures are easy to obtain and what structures are hard to make.
During training, a neural network assigns scores to reaction pairs (reactant → product) such that the product score must always be ≥ reactant score + a fixed margin. This means SCScore does not learn "a complexity metric" - it learns the chemical common sense that synthesis is a one-way journey toward increasing structural complexity.
Its scores are naturally aligned with real synthesis logic rather than just static structural observation.
Strengths: Grounded in real reaction data rather than expert-defined rules, making its scores naturally aligned with true synthesis logic. The directionality constraint gives the model a chemically meaningful training objective rather than an arbitrary complexity metric. Generalizes well across diverse chemical spaces.
Limitations: Does not generate explicit synthesis routes - it tells you how hard a molecule is likely to be to make, but not how to make it. Because the model is trained using historical reaction data, scores reflect what has been done before, which can underestimate the accessibility of genuinely novel but achievable compounds. Less interpretable than fragment-based methods, making it harder to pinpoint which structural features drive a high complexity score.
RAscore: A Fast Alternative to Full Retrosynthesis Planning
Running complete retrosynthetic analysis on millions of molecules in a virtual screen would be computationally prohibitive.
RAscore (Retrosynthetic Accessibility Score) was built specifically for efficiency.5 It is a binary classification model designed to quickly determine whether a molecule can have a complete retrosynthetic pathway planned by the professional tool AiZynthFinder.
Strengths: Far faster than full retrosynthetic analysis. Fully compatible with high-throughput screening. Simple to deploy without building a retrosynthesis engine.
Limitations: Only provides a binary yes/no judgment - no continuous difficulty score. Its ceiling capability is entirely determined by the underlying CASP tool.
DeepSA: Reading Molecules Like NLP Reads Text
DeepSA applies natural language processing (NLP) techniques to chemistry.6 It treats molecular SMILES strings as sentences, applies to pretrained language models, and outputs "easy to synthesize" or "hard to synthesize" labels along with a confidence score.
Importantly, its training labels are derived from the number of retrosynthetic steps computed by tools like Retro* - molecules requiring fewer than 10 steps are labeled easy; more than 10 steps or planning failure is labeled hard. It is not trained in real experimental data.
Therefore, DeepSA is learning whether an algorithm can plan a route, not whether a chemist can make the compound. This distinction matters in practice.
Strengths: Leverages pretrained molecular language models, capturing complex sequence-level patterns that fingerprint-based methods miss. Inference is fast once the model is loaded and scales well to large virtual libraries. Outputs a classification of confidence score alongside the binary label, giving some indication of model certainty.
Limitations: Labels are derived from algorithmic retrosynthesis step counts, not real experimental data, so the model learns what software can plan rather than what a chemist can execute in the lab. The binary output provides no granularity for ranking or prioritizing molecules within either class. Performance may degrade chemical spaces far removed from the training distribution, particularly for highly novel scaffolds.
GASA: Graph Neural Networks for Key Substructures
GASA (Graph Attention-based assessment of Synthetic Accessibility) models molecules as atom-bond graphs - atoms as nodes, chemical bonds as edges - and uses multi-head graph attention layers to automatically learn key structural features, incorporating bond-level information to strengthen global representation.7
Strengths: Strong generalization. Sensitive to subtle structural differences that lead to dramatically different synthesis difficulty. Excellent interpretability - atom-level contribution visualization shows chemists exactly which atoms and substructures drive synthesis difficulty.
Limitations: Training labels also depend on retrosynthetic tools like Retro*, introducing some gaps from real experimental outcomes. Graph attention computation is heavier than traditional fingerprint models, making it less ideal for ultra-large-scale screening.
ChemAIRS SA Score Super Fast Mode: Grounded in Real Synthesis Step Counts
ChemAIRS SA Score goes beyond traditional complexity-based metrics by incorporating real synthesis step counts, building block costs, and reaction feasibility. It was compared to deliver assessments that closely reflect how an experienced chemist would actually plan a route. The tradeoff for that depth has historically been speed.
That changes with ChemAIRS new SA Score Super Fast Mode. By rearchitecting the calculation engine, we've achieved an order-of-magnitude improvement in speed without compromising the core advantage that sets ChemAIRS apart: synthesis accessibility scores grounded in real, chemist-relevant synthesis data - not molecular complexity proxies.
What sets it apart:
Fully real-world data-driven. Trained and tested directly on the true shortest synthesis step counts of 1.5 million+ drug-like molecules. No handcrafted rules, no indirect predictions from retrosynthesis software - just real experimental data, which makes predictions naturally closer to what happens in the lab.
Rigorous time-split validation. Data before 2023 is used for training and validation; data from 2023 onward is held out for testing exclusively. This prevents data leakage and gives an honest picture of how the model generalizes to genuinely novel drug-like molecules.
End-to-end nonlinear learning. A neural network captures the complex, nonlinear relationship between molecular structure and synthesis step count. Unlike traditional models, ChemAIRS’ SA Score Super Fast Mode outputs a continuous score highly correlated with real synthesis steps - not a binary classification - giving medicinal chemists much richer decision support.
Integration of tens of millions of prior knowledge points. The model incorporates tens of millions of known molecules and commercially available starting materials, helping it avoid a common failure mode in other models: misjudging molecules that look structurally complex but have well-established synthesis routes.
Head-to-Head Performance
Systematic evaluation on an independent post-2023 test set showed that ChemAIRS SA Score Super Fast Mode achieved the highest Pearson correlation between predicted scores and actual synthesis step counts, demonstrating superior predictive accuracy. SCScore and the classic SAScore followed as strong second-tier performers.
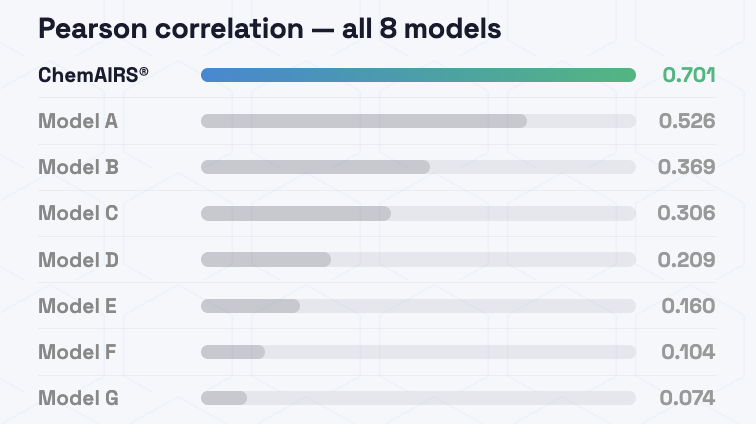
Figure 1: Pearson correlation between model predictions and ground truth synthesis steps
Virtual Screening Performance
In a more application-realistic virtual screening scenario - selecting 20,000 molecules for deep follow-up analysis - the results were:

ChemAIRS not only achieved the highest Top-20K selection precision, but also demonstrated strong stability - as the number of sampled molecules increases, its precision declines gradually rather than collapsing sharply. For real drug discovery projects, this stability is highly valuable.
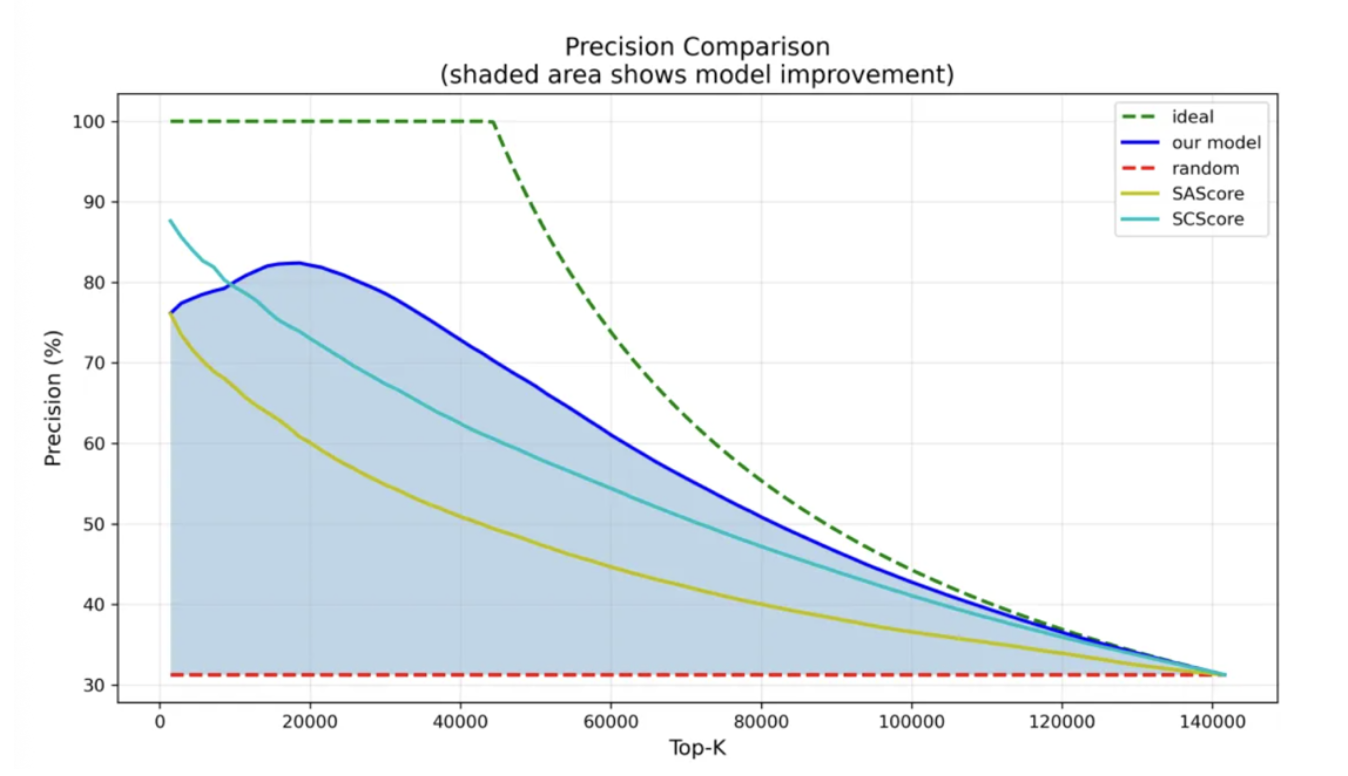
Figure 2: Prediction precision change across the top 10% of test set molecules
Conclusion
From SAScore's rule-based origins to SCScore's data-driven approach, and now to ChemAIRS's innovation grounded in real synthesis step counts - synthetic accessibility scoring has been steadily evolving over the past decade and a half.
ChemAIRS SA Score Super Fast Mode can evaluate 1 million molecules in approximately 30 minutes, rapidly filtering for synthesizable candidates. That said, a Pearson correlation of 0.7 is not perfect - some molecules that score well may still present challenges in the lab.
For scenarios requiring higher accuracy and interpretability, ChemAIRS's full retrosynthetic reasoning modes are recommended. These provide more rigorous synthesis feasibility assessment, stronger explainability, and concrete synthesis route suggestions for medicinal chemists.
Every advance in synthetic accessibility assessment chips away at the barrier between AI-generated molecules and experimental synthesis - steadily pushing drug discovery toward a faster, smarter future.
References
Ertl, P.; Schuffenhauer, A. Estimation of Synthetic Accessibility Score of Drug-like Molecules Based on Molecular Complexity and Fragment Contributions. J. Cheminform. 2009, 1, No. 8. doi:10.1186/1758-2946-1-8
Chen, S.; Jung, Y. Estimating the Synthetic Accessibility of Molecules with Building Block and Reaction-Aware SAScore. J. Cheminform. 2024, 16, No. 83, doi:10.1186/s13321-024-00879-0
Voršilák, M.; Kolář, M.; Čmelo, I.; Svozil, D. SYBA: Bayesian Estimation of Synthetic Accessibility of Organic Compounds. J. Cheminform. 2020, 12, No. 35. doi:10.1186/s13321-020-00439-2
Coley, C. W.; Rogers, L.; Green, W. H.; Jensen, K. F. SCScore: Synthetic Complexity Learned from a Reaction Corpus. J. Chem. Inf. Model. 2018, 58, 252-261. doi:10.1021/acs.jcim.7b00622
Thakkar, A.; Chadimová, V.; Bjerrum, E. J.; Engkvist, O.; Reymond, J.-L. Retrosynthetic Accessibility Score (RAscore) - Rapid Machine Learned Synthesizability Classification from AI Driven Synthetic Planning. Chem. Sci. 2021, 12, 3339-3349. doi:10.1039/D0SC05401A
Wang, S.; Wang, L.; Li, F.; Bai, F. DeepSA: A Deep-learning Driven Predictor of Compound Synthesis Accessibility. J. Cheminform. 2023, 15, No. 103. doi:10.1186/s13321-023-00771-3
Yu, J.; Wang, J.; Zhao, H.; Gao, J.; Kang, Y.; Cao, D.; Wang, Z.; Hou, T. Organic Compound Synthetic Accessibility Prediction Based on the Graph Attention Mechanism. J. Chem. Inf. Model. 2022, 62, 2973-2986. doi:10.1021/acs.jcim.2c00038